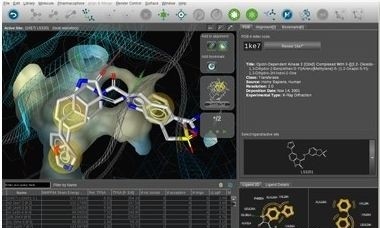
LigandScout is a software tool that allows to rapidly and transparently derive 3D pharmacophores from structural data of macromolecule/ligand complexes in a fully automated and convenient way.
The algorithms are scientifically published and based on several years of experience in pharmacophore creation, while the application corresponds to state-of-the-art information technology. Support for various common pharmacophore formats like the export to Catalysttm, MOEtm or Phasetm guarantee maximum interoperability to screening platforms.
The full-featured 3D graphical user interface with multiple undo-levels will make the exploration of the active site and pharmacophore creation within the complex transparent and efficient.
Binding site analysis, pharmacophore-based alignment and the creation of shared feature pharmacophores are designed to make LigandScout an essential tool for structure-based drug design in combination with virtual screening. LigandScout runs on all common operating systems.
Here are some key features of "LigandScout":
Structure-based 3D pharmacophore design:
· ligandscout screen shotAutomatic interpretation of PDB ligands using geometry, dictionaries and rules
· State-of-the-art user interface with advanced 3D graphics and undo-function
· 2D view and hierarchical view directly linked to 3D interface
· Fast alignment of molecules in their bio-active conformation to other molecules and 3D pharmacophores from several ligands and/or pharmacophores, shared feature pharmacophores can be derived to understand and model the relevant mode of action
· Advanced handling of co-factors, ions, water molecules and covalently bound ligands
· Pharmacophore export to Catalyst(tm), MOE(tm) and Phase(tm) for virtual screening
· Extensive parameter control for more experienced users
· Advanced PDB ligand perception and easy manual correction while modeling in the active site
· Ability to treat co-factors and water molecules as part of the ligand or part of the macromolecule
· Intuitive pharmacophore-based alignment of molecules
· Support for most common file formats
· Sophisticated file and repository management of edited and stored binding sites, molecules and pharmacophores
· Smart enumeration of tautomers
Accurate and fast virtual screening:
· High performance accurate virtual screening with automated analysis of screening performance using ROC curves and enrichment factor calculations
· Simple workflows for boolean combination of target and anti-target pharmacophore models
· Accurate implementation of the MMFF94 force field for generating high quality geometries
Ligand-based pharmacophore design:
· Ligand-based pharmacophore modeling, including automatic classification of chemical features, feature weights and generation of exclusion volume spheres
· Automated training set selection by pharmacophore- based cluster analysis
Requirements:
· at least 2GB of RAM (4 GB recommended)
· Hardware-accelerated 3D graphics card (nvidia or ATI recommended) with OpenGL 1.2 support
Limitations:
· 30 days trial